Winmostar 計算メニュー
MOPAC キーワード | 分子軌道表示| Energy Leve|Charge, Dipole | Animation| IRC, STEP|Force
GAMESS キーワード| Animation| MO, Charge, Dipole|振動スペクトル
Gaussian キーワード|Animation| 最適化構造|MO, UV, Charge, Dipole|振動スペクトル| Fchk (Cubegen)| Cube Plot
CNDO/S キーワード|UV-VIS スペクトル表示
MOPAC
MOPAC キーワード Setup
MOPACの計算を実行するためのデータファイルに記述されるキーワードのリストのデフォルトを設定します。
各項目を選択して指定し、[Set]ボタンをクリックします。
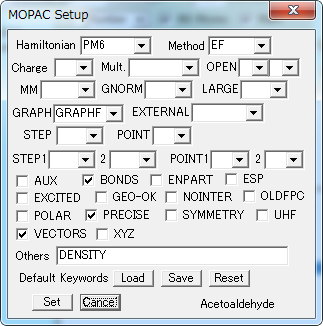
| Hamiltonian | 使用するハミルトニアンを指定します。 |
| AM1 | MOPAC 6, MOPAC 7, MOPAC 93, MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006, MOPAC 2009 |
| PM3 | MOPAC 6, MOPAC 7, MOPAC 93, MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006, MOPAC 2009 |
| RM1 | MOPAC 2009 |
| AM1 EXTERNAL=RM1.rm1 | MOPAC 7.1, MOPAC 93, MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006, MOPAC 2009 |
| PM5 | MOPAC 2002, MOPAC 2006 |
| PM6 | MOPAC 2009 |
| MINDO/3 | MOPAC 6, MOPAC 7, MOPAC 93, MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006 |
| MNDO | MOPAC 6, MOPAC 7, MOPAC 93, MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006, MOPAC 2009 |
| MNDO-d | MOPAC 97, MOPAC 2000, MOPAC 2002, MOPAC 2006, MOPAC 2009 |
| Method | 計算方法を指定します。 |
| EF | EF (Eigen Vector Following)法による構造最適化計算を行います。 |
| TS | 遷移状態を求めます。 |
| FORCE | 振動解析を行います。 |
| 1SCF | 1回だけSCF計算を行います。(構造最適化を行いません。) |
| IRC | 固有反応座標計算を行います。エネルギーは保存されません。 |
| IRC=1 | 1番目の基準振動の逆方向を指定して固有反応座標計算を行います。 |
| IRC=-1 | 1番目の基準振動の正方向を指定して固有反応座標計算を行います。 |
| Charge | 電荷の値を指定します。 |
| Multiplicity | 多重度を指定します。 |
| OPEN | 開殻計算における電子数と軌道数を指定します。 |
| MM | MMOK: CONH結合に分子力学補正を加えます。 NOMM: CONH結合に分子力学補正を加えません。 |
| GNORM | エネルギー勾配ノルムの閾値を指定します。 |
| LARGE | 指定したサイクルごとに情報を出力します。 |
| GRAPH | 分子軌道をグラフィックス表示するためのファイルを作成します。(GPAGH/GRAPHF) |
| EXTERNAL | ディスク上のパラメータ・ファイルを読み込みます。 |
| STEP | 反応座標計算におけるきざみ幅を指定します。 |
| POINT | 反応座標計算における計算点数を指定します。 |
| STEP1/2 | グリッド計算におけるきざみ幅を指定します。 |
| POINT1/2 | グリッド計算における計算点数を指定します。 |
| BONDS | 最終の結合次数行列を出力します。 |
| ENPART | エネルギーを1中心および2中心項に分解するエネルギー分割を指定します。 |
| ESP | 静電ポテンシャルを計算します。 |
| EXCITED | 一重項第一励起状態を最適化します。 |
| GEO-OK | 原子が異常に近接している場合のチェックを無視します。 |
| NOINTER | 原子間距離を出力しません。 |
| OLDFPC | 古いバージョンのMOPACと同じ基準物理量の値を用います。 |
| POLAR | 分極率を計算します。 |
| PRECISE | 収束判定条件を 100 倍厳しくします。 |
| SYMMETRY | 対称性や等価条件を利用して構造を定義します。 |
| UHF | 非制限Hartree-Fock計算を実行します。 |
| VECTORS | 最終固有ベクトル(波動関数)を出力します。 |
| XYZ | XYZ座標系を用いて計算を行います。 |
| Others | その他のキーワードを記入します。 |
| Default Keywords | |
| Load | 保存してある設定を読み込みます。 |
| Save | 設定を保存します。 |
| Reset | 初期化します。 |
| Set | 設定します。 |
| Cancel | 設定を行わずに終了します。 |
Mopac 分子軌道表示 (mgf, gpt)
MOAPCによる計算結果にもとづいて分子軌道を図示します。
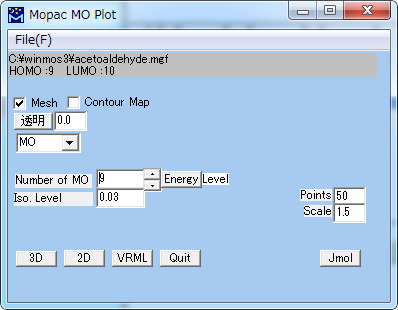
| Mesh | 分子軌道の等値面を格子で表現します。 |
| Contour Map | 分子軌道の等値面をソリッドモデルで表現します。 |
| 透明 | 透明度を指定します。(0: 不透明、1: 透明) |
| Number of MO | 表示する分子軌道の番号を指定します。 (HOMOとLUMOの番号が上部に表示されています。) |
| Iso. Level | 描く確率電子密度の等値面の値を指定します。 (1/A^3) |
| Points | 格子点の数 (デフォールト 50) |
| Scale | 描く範囲を指定するスケール・ファクター (デフォールト 1.5) |
| EnergyLevel | エネルギー準位の図を表示します。 |
| 3D | 3D Viewerを利用して表示します。 |
| 2D | 分子表示ウィンドウ内で格子で表現します。 |
| VRML | VRMLで表示します。(VRMLビューワがインストールされている必要があります) |
| Quit | このウィンドウを閉じます。 |
| Jmol | Jmol用のスクリプトを出力します。 |
| ※ 3D表示の際のメニューについては、3D Viewer を参照してください。 | |
| ※ 3D表示の際のPreferenceについては、3D - Preferenceを参照してください。 | |
Energy Level Diagram
分子軌道のエネルギー準位を表示します。
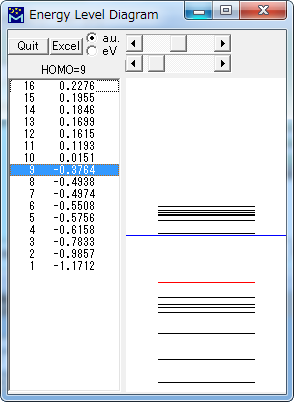
| Excel | Excelを起動し、エネルギー順位の表を出力します。 |
| a.u./eV | 単位を切り替えます。 |
| スライダー | 原点位置を移動します。 |
| 拡大率(描画範囲)を指定します。 | |
| Quit | ウィンドウを閉じます。 |
MOPAC Import Charge, Dipole (arc)
MOPACによる計算の結果を読み込んで、双極子モーメントを表示します。
「表示」−「双極子モーメント」メニューで、「表示」にチェックされていることが必要です。
MOPAC Import Animation (arc)
MOPACによるMinimum Energy Path(Reaction Coordinate)計算の結果を読み込んで、アニメーションを表示します。
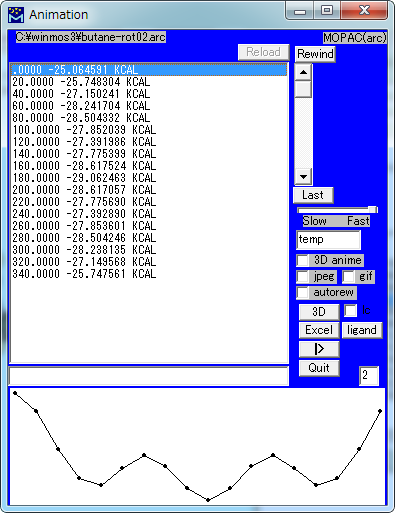
| Animation | |
| スライダー | フレームを移動します。 |
| Rewind | 最初のフレームに移動します。 |
| Last | 最後のフレームに移動します。 |
| Slow - Fast | 速度を設定します。 |
| ファイル名 | 書き出すファイル名を指定します。 |
| 3D anime | 3D Viewer画面でアニメーションを表示します。 |
| Jpeg | アニメーションを実行する際に一連のJPEGファイルを書き出します。 |
| GIF | アニメーションを実行する際に一連のGIFファイルを書き出します。 |
| autorew | 自動的に巻き戻して連続してアニメーションを実行します。 |
| 3D | 3D Viewerを起動して表示し、アニメーションの設定ウィンドウを表示します。 |
| Excel | CSVファイルを出力し、Excelを起動して読み込みます。 |
| ligand | CSVファイルを出力し、Excelを起動して読み込みます。 |
| >| ( | ) | アニメーションの実行 (停止) |
| Quit | Animation ウィンドウを閉じます。 |
MOPAC Import IRC, STEP (out)
MOPACによるIRC計算結果を読み込んで、アニメーションを表示します。
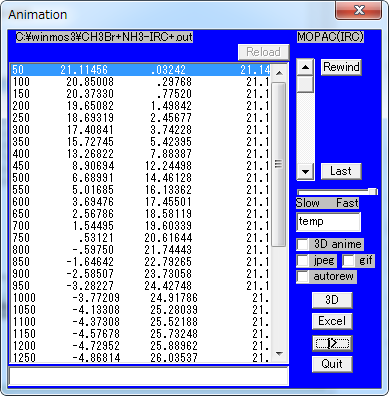
| Animation | |
| スライダー | フレームを移動します。 |
| Rewind | 最初のフレームに移動します。 |
| Last | 最後のフレームに移動します。 |
| Slow - Fast | 速度を設定します。 |
| 3D anime | 3D Viewer画面でアニメーションを表示します。 |
| Jpeg | アニメーションを実行する際に一連のJPEGファイルを書き出します。 |
| GIF | アニメーションを実行する際に一連のGIFファイルを書き出します。 |
| 3D | 3D Viewerを起動して表示し、アニメーションの設定ウィンドウを表示します。 |
| Excel | CSVファイルを出力し、Excelを起動して読み込みます。 |
| >| ( | ) | アニメーションの実行 (停止) |
| Quit | Animation ウィンドウを閉じます。 |
MOPAC Import Force (out)
MOPACによるForce計算結果を読み込んで、振動スペクトルを表示します。
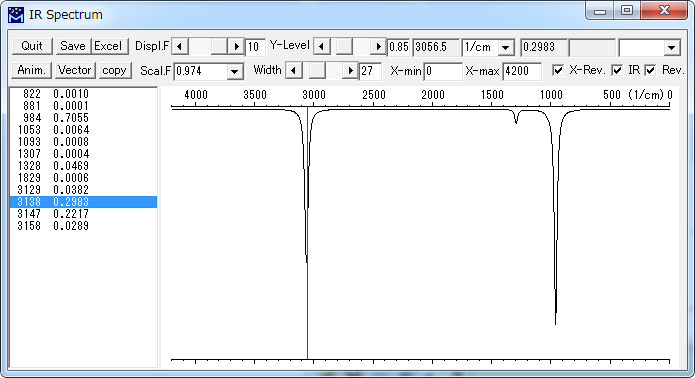
左側の一覧からクリックして選択したピークの位置が赤線で表示されます。
| IR Spectrum | |
| Quit | IR Spectrum ウィンドウを閉じます。 |
| Save | スペクトルの図をGIFファイルまたはJPEGファイルを書き出します。 |
| Excel | CSVファイルを出力し、Excelを起動して読み込みます。 |
| Anim. | 選択された振動数(ピーク)に相当する振動のアニメーション表示を行います。 |
| Vector | 選択された振動数(ピーク)に相当する振動のベクトル表示を行います。 |
| copy | スペクトルの図をクリップボードにコピーします。 |
| Displ.F | |
| Y-Level | 縦軸の縮尺を変更します。 |
| 単位 | 1/cm、eV、nmから単位を選択します。 |
| Scal.F. | 系統的誤差を補正するためのスケーリング・ファクターを選択します。 |
| Width | 半値幅を設定します。 |
| X-min | 波数(波長)軸の表示する範囲を指定します。 |
| X-max | 波数(波長)軸の表示する範囲を指定します。 |
| X-rev | 波数(波長)軸を反転して表示します。 |
| IR | IRスペクトルを表示します。 |
| Rev. | 上下を反転して表示します。(吸収スペクトル) |
GAMESSp
GAMESS キーワード Setup
GAMESSによる計算を実行するためのデータファイルに記述されるキーワードなどを設定します。
各項目を選択して指定し、[Set]ボタンをクリックします。
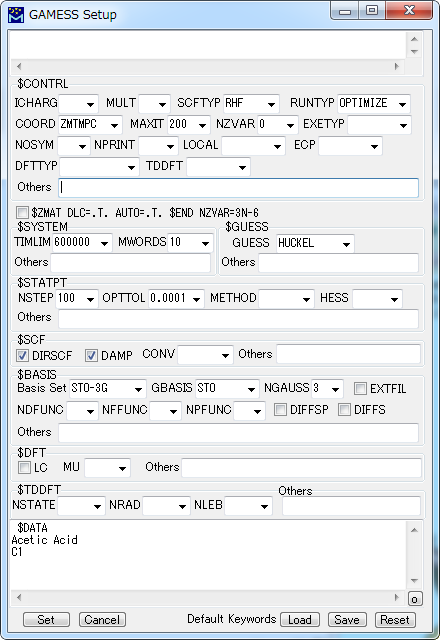
| ICHARG | 電荷を指定します。 |
| MULT | 多重度を指定します。 |
| SCFTYP | SCF計算方法を指定します。 |
| RUNTYP | 計算目的を選択します。 |
| COORD | 分子構造データの形式を指定します。 |
| MAXIT | SCF計算の反復回数の上限を指定します。 |
| NZVAR | 内部座標の数を指定します。 |
| EXETYP | 実際に計算を行うかどうかの指定で、入力をチェックするときはCHECKを指定します。 |
| NOSYM | 計算の際に対称性を利用するかどうかを指定します。 |
| NPRINT | 出力の詳細度を指定します。 |
| LOCAL | 軌道の局在化の方法を指定します。(デフォルト 0 = しない) |
| ECP | Pseudopotentialを指定します。 |
| DFTTYP | 密度汎関数法の基底関数系を指定します。 |
| TDDFT | 時間依存(Time-dependent)DFT法を用いて励起状態のエネルギー計算を行うかどうかを指定します |
| Others | その他のキーワードを記入します。 |
| $ZMAT DLC=.T. AUTO=.T. $END NZVAR=3N-6 | |
| &SYSTEM | |
| TIMLIM | 計算の制限時間 (デフォルト 600分) |
| MWORDS | メモリー使用量 (デフォルト 1MW) |
| Others | その他のキーワードを記入します。 |
| $GUESS | |
| GUESS | 初期波動関数の求め方を指定します。 |
| Others | その他のキーワードを記入します。 |
| $STATPT | |
| NSTEP | 構造最適化のステップ数の上限を指定します。(デフォルト 20) |
| OPTTOL | エネルギー勾配の閾値を指定します。(デフォルト 0.0001 Hartree/Bohr) |
| METHOD | 構造最適化のアルゴリズムを指定します。 |
| HESS | Hessian 行列の求め方を指定します。 |
| Others | その他のキーワードを記入します。 |
| $SCF | |
| DIRSCF | ダイレクトSCF計算法を使用するかどうかを指定します。 |
| DAMP | Fock 行列の作成に際して、Davidson damping を利用します。 |
| CONV | SCF収束判定の際の密度変化の閾値を指定します。(デフォルト 1.0D-05) |
| Others | その他のキーワードを記入します。 |
| $BASIS | |
| Basis Set | 基底関数系を選択します。 |
| GBASIS | 基底関数系の基本セット |
| NGAUSS | Gaussian関数の数 |
| NDFUNC | 加えるd-分極関数の数 |
| NFFUNC | 加えるf-分極関数の数 |
| NPFUNC | 加えるp-分極関数の数 |
| DIFFSP | sp-diffuse関数を加えるかどうかの指定 |
| DIFFS | s-diffuse関数を加えるかどうかの指定 |
| EXTFIL | 外部ファイルから基底関数を読み込みます。 |
| Others | その他のキーワードを記入します。 |
| $DFT | |
| LC | 遠距離補正を行うかどうかを指定します。(BLYP, BOP及び BVWNの場合のみ) |
| MU | 遠距離補正のパラメータの値を指定します。(デフォルト 0.33) |
| Others | その他のキーワードを記入します。 |
| $TDDFT | |
| NSTATE | 求める状態の数(基底状態をのぞく)を指定します。 |
| NRAD | 密度汎関数の導関数を求める際の動径方向の格子点の数を指定します。(デフォルト 48) |
| NLEB | 角度方向の格子点の数を指定します。(デフォルト 110) |
| Others | その他のキーワードを記入します。 |
| Default Keywords | |
| Load | 保存してある設定を読み込みます。 |
| Save | 設定を保存します。 |
| Reset | 初期化します。 |
| Set | 設定します。 |
| Cancel | 設定を行わずに終了します。 |
GAMMES Import
Animation
GAMMESによる計算結果にもとづいてアニメーションを図示します。
※ メニューについては、MOPAC IRC Out の項を参照してください。
戻 る
MO, Charge, Dipole(分子軌道と電荷などの表示)
GAMMESによる計算結果にもとづいて分子軌道と電荷などを図示します。
※ 分子軌道の表示メニューについては、MOPAC MO Plot の項を参照してください。
振動スペクトルの表示 (Hessian/Raman)
GAMMESによる計算結果にもとづいて振動スペクトルを図示します。
※ メニューについては、MOPAC Import Force (out)の項を参照してください。
Gaussian
Gaussian キーワード Setup
Gaussianによる計算を実行するためのデータファイルに記述されるキーワードのリストのデフォルトを設定します。
各項目を選択して指定し、[Set]ボタンをクリックします。
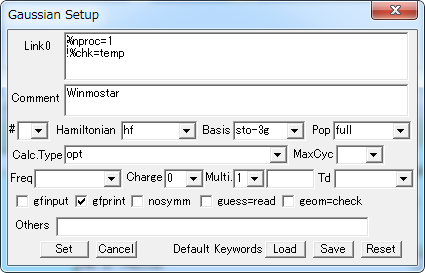
| Link0 | |
| #nproc=n | プロセッサ数を指定します。 |
| #Chk=file | チェックポイントファイルを指定します。 |
| #Mem=n | 動的メモリ量をワード単位で指定します。KB, MB, GB, KW, MB, GWの単位を指定することもできます。(デフォルト:6MW) |
| Comment | コメントを記述します。 |
| # | ルートセクションの始まりを指定します。 |
| #N | 標準レベルで出力を行います。(デフォルト) |
| #P | 詳細な出力を行います。各リンクの開始時と終了時における実行時間などや,SCFの収束に関する情報が出力されます。 |
| #T | 重要な情報と結果のみを出力する簡潔な出力を指定します。 |
| Hamiltonian | 使用するハミルトニアンを指定します。 |
| hf | Hartree-Fock計算を行います。明示的に指定されない限り,一重項にはRHFを,それより高次の多重度ではUHFを用います。 |
| rhf | Restricted Hartree-Fock計算を行います。 |
| uhf | Unrestricted Hartree-Fock計算を行います。 |
| am1 | AM1ハミルトニアン を用いた半経験的計算を行います。 |
| pm3 | PM3ハミルトニアン を用いた半経験的計算を行います。 |
| pm3mm | HCON結合に関する分子力学補正が含まれたPM3ハミルトニアン を用いた半経験的計算を行います。 |
| b3lyp | Becke3汎関数にLYP非局所相関汎関数を組み合わせた密度汎関数法計算を行います。 |
| ub3lyp | 開殻系のための非制限B3LYP法 |
| mp2 | Hartree-Fock計算の後に2次までのMoller-Plesset相関エネルギー補正を行います。 |
| ump2 | 開殻系のための非制限MP2法 |
| mp4 | Hartree-Fock計算の後に4次までのMoller-Plesset相関エネルギー補正を行います。 |
| ump4 | 開殻系のための非制限MP4法 |
| cis | 1電子励起配置間相互作用計算を行います。 |
| cisd | 1および2電子励起配置間相互作用計算を行います。(CIと同義) |
| indo | INDOハミルトニアンを用いた半経験的計算を行います。 |
| zindo | ZernerのINDO/S法による光吸収の計算を行います。 |
| zindo(nstates=10) | ZernerのINDO/S法による光吸収の計算を行い、10の励起状態に対する解を求めます。 |
| gvb | GVB(General Valence Bond; 一般化原子価結合)計算を行います。 |
| oniom | ONIOM計算を行います。 |
| Basis | 基底関数セットを指定します。 |
| Pop | 分子軌道や電子密度解析及び原子の電荷分布などの出力を制御します。 |
| none | 分子軌道を出力せず,電子密度解析も行いません。 |
| minimal | 原子の電荷と軌道エネルギーを出力します。 |
| regular | 占有軌道と仮想軌道を5つずつ出力します。密度行列とMulliken電子密度解析も出力します。 |
| full | すべての占有軌道と仮想軌道を出力します。密度行列とMulliken電子密度解析も出力します。 |
| Calc. Type | |
| opt | 構造最適化を実行します。 |
| opt=z-matrix | 内部座標で構造最適化を行います。 |
| opt=modredundant | redundant内部座標の定義(探索や束縛情報を含む)を追加・削除・修正します。構造指定部の後に入力セクションが必要です。 |
| opt=(ts,noeigentest,calcfc) | 遷移状態に対する最適化を行います。曲率のテストを行いません。初回に力の定数を計算します |
| irc | 反応経路を追跡します |
| irc=(maxpoint=20,stepsize=20t,calcfc) | 反応経路を追跡します。経路上の点の個数とステップサイズを指定します。初回に力の定数を計算します |
| MaxCyc=n | 最適化ステップの最大数をnにします。 |
| Charge | 電荷の値を指定します。 |
| Multi. | 多重度を指定します。 |
| Freq | |
| freq | 力の定数と振動数の計算を行います。 |
| freq=raman | IR強度に加えてラマン強度も計算します。 |
| freq=vcd | 通常の振動数解析に加えて振動円二色性(VCD)強度を計算します |
| freq=noraman | Hartree-Fock解析的振動数計算でラマン強度を求めません。 |
| freq=nraman | 電場に関する解析的双極子導関数を数値的に微分することによって分極率導関数を求めます。 |
| freq=nnraman | 核座標に関する解析的分極率を数値微分して分極率導関数を求めます。 |
| Td | |
| td | 時間依存(time-dependent)Hartree-FockまたはDFT法を用いて励起状態のエネルギー計算を行います |
| td=(nstates=n) | n個の状態に対して時間依存計算法を用いて励起状態のエネルギーを求めます。(デフォルト 3) |
| gfinput | 基底関数系を入力フォーマットと同様な形式で出力します。 |
| gfprint | 基底関数系を表形式で出力します。 |
| nosym | 座標の再配向を行わず、Z-matrix 配向ですべての計算を実行します。 |
| guess=read | チェックポイントファイルから初期波動関数を読み込みます |
| geom=check | 分子構造指定セクションをチェックポイントファイルから取り出します。 |
| Others | その他のキーワードを記入します。 |
| Default Keywords | |
| Load | 保存してある設定を読み込みます。 |
| Save | 設定を保存します。 |
| Reset | 初期化します。 |
| Set | 設定します。 |
| Cancel | 設定を行わずに終了します。 |
Gaussian Import Animation
Gaussianによる計算結果にもとづいてアニメーションを図示します。
※ メニューについては、MOPAC IRC Out の項を参照してください。
Gaussian Import Opt
Gaussianによる計算結果にもとづいて最適化構造を図示します。
※ メニューについては、MOPAC Import IRC, STEP (out) の項を参照してください。
Gaussian Import MO, UV, Charge, Dipole (分子軌道と紫外・可視スペクトル及び電荷などの表示)
Gaussianによる計算結果にもとづいて分子軌道と紫外・可視スペクトル及び電荷などを図示します。
※ 分子軌道の表示メニューについては、MOPAC Import - MO (mgf, gpt) の項を参照してください。
※ 紫外・可視スペクトルの表示メニューについては、UV-VISスペクトル表示 の項を参照してください。
Gaussian Import Freq 振動スペクトルの表示
Gaussianによる計算結果にもとづいて振動スペクトルを図示します。
※ メニューについては、MOPAC Import Force (out)の項を参照してください。
Gaussian Import Fchk (Cubegen)
Gaussian for Windows ユーティリティのCubegenを起動し、.fchファイルを読込んでCubeファイルを作成します。
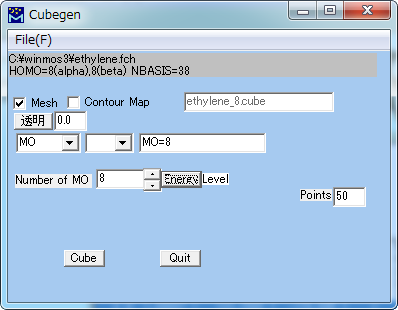
| Mesh | 分子軌道の等値面を格子で表現します。 |
| Contour Map | 分子軌道の等値面をソリッドモデルで表現します。 |
| 透明 | 透明度を指定します。(0: 不透明、1: 透明) |
| Property | |
| / MO | 分子軌道 |
| / Density | 電子密度 |
| / ESP | スピン密度 |
| / Spin | スピン密度(α - β) |
| / Alpha | αスピン密度 |
| / Beta | βスピン密度 |
| / Current Density | 磁場存在下(GIAO)での電子密度の大きさ |
| / Shielding Density | 磁気遮蔽密度 |
| Type | Density キーワードのオプションを指定します。 (HF, MP2, CI, QCI) |
| Number of MO | 対象とするする分子軌道の番号を指定します。 (HOMOの番号が上部に表示されています。) |
| Points | 一辺あたりの格子点数を指定します。 |
| Cube | Cubeファイルを出力します。 |
| Quit | このウィンドウを閉じます。 |
Gaussian Import Cube (Gaussian Cube Plot)
Cubeファイルを読込んで図示します。
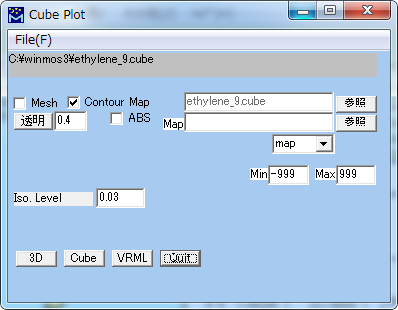
| File | 「ファイルを開く」ダイアログを表示し、cubeファイルを読み込みます。 |
| Mesh | 分子軌道の等値面を格子で表現します。 |
| Contour Map | 分子軌道の等値面をソリッドモデルで表現します。 |
| 透明 | 透明度を指定します。(0: 不透明、1: 透明) |
| ABS | |
| 参照 | 「ファイルを開く」ダイアログを表示し、cubeファイルを読み込みます。 |
| Map | |
| / 参照 | 「ファイルを開く」ダイアログを表示し、マッピングに用いる2番目のcubeファイルを読み込みます。 |
| / map | 上の欄のデータに下の欄のデータをマッピングします。(例 DensityにESPをマッピングする) |
| / subtract | 2つのcubeファイルのデータの差を対象とします。 |
| / sub 2 | 2つのcubeファイルのデータの自乗の差を対象とします。 |
| / add | 2つのcubeファイルの和を対象とします。 |
| Iso. Level | 描く確率電子密度の等値面の値を指定します。 (1/A^3) |
| Scale | 描く範囲を指定するスケール・ファクター (デフォールト 1.5) |
| 3D | 3D Viewerを利用して表示します。 |
| Cube | Mapで対象としたcubeファイルの演算結果を出力し表示の対象とします。 |
| VRML | VRMLで表示します。(VRMLビューワがインストールされている必要があります) |
| Quit | このウィンドウを閉じます。 |
| ※ 3D表示の際のメニューについては、3D Viewer を参照してください。 | |
| ※ 3D表示の際のPreferenceについては、3D - Preferenceを参照してください。 | |
UNIX Server
Submit Job
UNIXサーバ上のGaussinaプログラムによる計算ジョブを実行するための設定をします。
| Profile | 登録されているプロフィールから設定を選択します。 |
| New | 新しいプロフィールを開きます。 |
| Copy | Profile欄に表示されているプロフィールをコピーします。 |
| Rename | プロフィールの名前を変更します。OKをクリックすると確定します。 |
| Delete | Profile欄に表示されているプロフィールを削除します。 |
| Quit | このウィンドウを閉じます。 |
CNDO/S
CNDO/S キーワード Setup
CNDO/S法による計算を実行するための設定を行います。
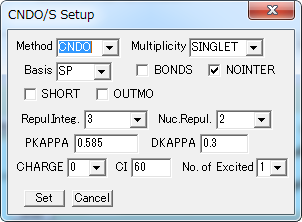
| Method | 計算方法を指定します。 (CNDO または INDO) |
| Multiplicity | 一重項(または三重項)励起状態の計算を指定します。 SINGLET (TRIPLET) |
| Basis | 基底関数を指定します。(SP または SPD) |
| BONDS | 結合次数を出力することを指定します |
| NOINTER | 原子間距離を出力しません。 |
| SHORT | 出力の簡略化を指定します。 |
| OUTMO | MOLMOL2用のファィルを出力します。 |
| Repul.Integ. | 反発積分の式を指定します。 1: Pariser 2: 大野 3: 西本−又賀 4: 理論式 |
| Nuc.Repul. | 核間反発エネルギーの式を指定します。 1: Za * Zb / 1 2: Za * Zb * γab |
| PKAPPA | p電子に対するΚの値を指定します 。 デフォルト値 0.585 |
| DKAPPA | d電子に対するΚの値を指定します。 デフォルト値 0.3 |
| CHARGE | 電荷 |
| CI | 励起状態のCI 計算に含める状態の数を指定します。 (上限 500) |
| No. of Excited | 結合次数を出力する励起状態の数を指定します。 |
| Set | 設定します。 |
| Cancel | 設定を行わずに終了します。 |
| 原子種、座標 | Winmostarが自動的に生成します。 |
UV-VIS スペクトル表示
Gaussian、CNDO/S及びMOSFによる計算結果にもとづいて紫外・可視スペクトルの吸収位置、強度の理論値を図示します。
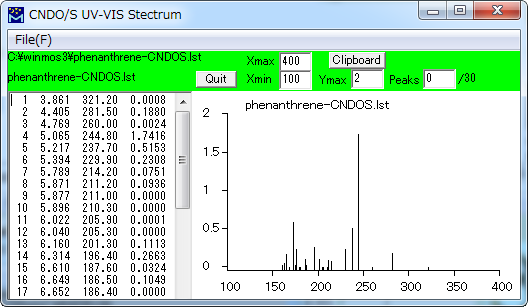
| File | |
| Open | CNDO/SまたはMOSFによる出力ファイルを読み込みます。 |
| Exit | ウィンドウを閉じます。 |
| Quit | ウィンドウを閉じます。 |
| Xmax | 表示する波長の上限 (nm) |
| Xmin | 表示する波長の下限 (nm) |
| Ymax | 縦軸(振動子強度)の上限 |
| Peaks | 指定した数のピークについて波長と振動子強度の値を図の中に記入します。(長波長側から) |
| ※ Xmax、Xmin、Ymax、Peaksの指定は、 ウィンドウサイズを変更した際に反映されます。 | |
※ 「MO Plot」ウィンドウメニューについては、MOPAC Import MO (mgf, gpt) の項を参照してください。