Basic Operation of JSmol
Mouse Operation
Every action cna be achieved with one, two and three button mouse.
| Function | Main Button (Left) | Middle Button | Secondary Button (Right) |
|---|---|---|---|
| Open JSmol Menu | click on Jmol logo or hold Ctrl while clicking | click | |
| Rotate around X, Y | drag | ||
| Move along X, Y | hold Shift while double-clicking and holding down on the second click for dragging | double-click and holding down on the second click for dragging | hold Ctrl while dragging |
| Rotate around Z | hold Shift while dragging horizontally | drag horizontally | hold Shift while dragging horizontally (may fall on Mac's) |
| Zoom | hold Shift while dragging vertically or use mouse wheel | drag vertically | |
| Reset | hold Shift while double-clicking away from the molecule | double-click away from the molecule |
Reference Web address
Refernce guide for Jmol command and scripting language : http://chemapps.stolaf.edu/jmol/docs/
Jmol web site : http://jmol.sourceforge.net
Selection of Schemes
Radio Button Controls
You can use the radio button controls shown at the foot of the applet window.
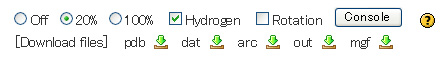
Atoms are rendered as spheres based at the size of van der Waals radius of each element.
"Off" corresponds to "Stick Model", "20%"corresponds to "Ball and Stick Model" and "100%" corresponds to "CPK (Spacefill) Model".
Jmol Menu
Jmol menu offers a wide assortment of options. Scheme is set using Style menu.
Style > Scheme > CPK Spacefill (Ball and Stick, Sticks, Wireframe)
In the case of proteins, the menu to use is Structures.
Style > Structures > Backbone (Off, Cartoon, Cartoon Rockets, Ribbons, Rockets, Strand, Trace)
Measurement of Distances and Angles
Distance (2 atoms):
- double-click on the starting atom
- to fix a distance measurement, double-click on second atom
Angle (3 atoms):
- double-click on the starting atom
- click on the second atom (central atom in angle)
- to fix an angle measurement, double-click on third atom
Torsion angle or dihedral angle (4 atoms)
- double-click on the starting atom
- click on the second atom
- click on the third atom
- to fix a dihedral angle measurement, double-click on fourth atom
Make the same measurement again in order to delete the measurement.
Measurement of Distances and Angles Without Leaving a Permanent Measurement
Move pointer over destination atom in order to see measurement results without leaving a permanent measurement.
Click away from the molecule in order to cancel the measurement.
Slab - Cutting plane
Open the script editor window. Type "slab on" and click the "Run" button.
| Function | Main button (Left) |
|---|---|
| Slab (slab from front) | hold Ctrl + Shift while dragging vertically |
| Depth (slab from back) | hold Ctrl + Shift while double-clicking and holding down on the second click for dragging vertically |
| Shift the slab | hold Alt + Ctrl + Shift while dragging vertically |
Example