MOPACの使用例 − スチレンのモデリングとMOPAC計算
スチレンの分子構造の構築
初期画面で、分子表示ウィンドウの上部にある[-C6H5]をクリックしてフェニル基を指定した後、[Rep]をクリックするか炭素原子を右クリックします。
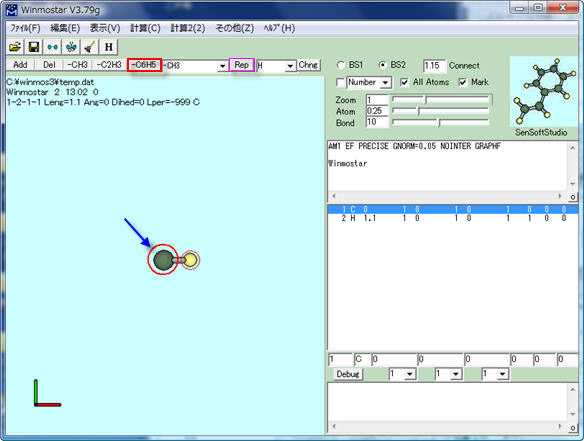
ベンゼンの構造ができました。
次に、分子表示ウィンドウの上部にある[-C2H3]をクリックしてビニル基を指定した後、[Rep]をクリックするか接続したい位置の水素原子を右クリックします。
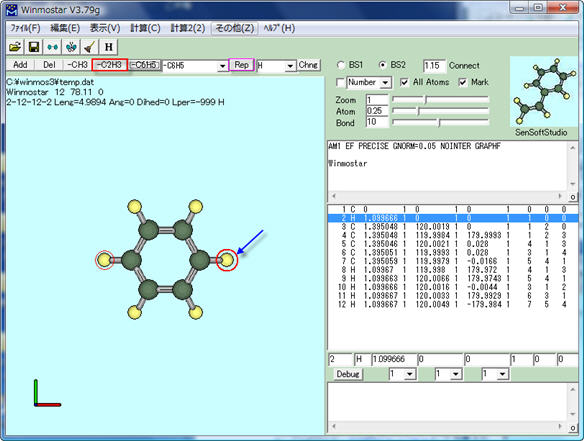
スチレンの構造ができました。
MOPACによる分子軌道法計算の実行
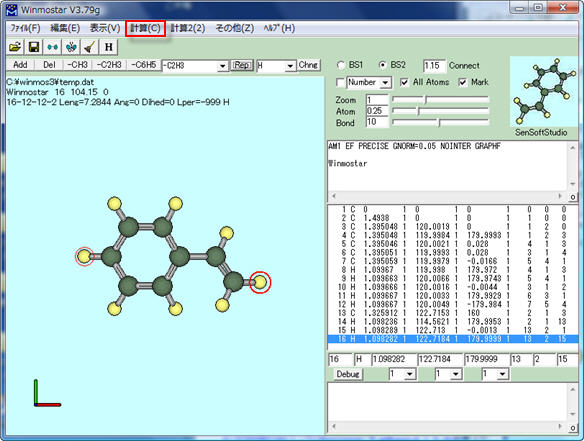
[計算]メニューから[(1)MOP6W70 Start] (または[(2)MOP7W70 Start]あるいは[(3)MOPAC 2009 Start]) を選びます。
分子が編集されたままファイルの保存が行われていないので、構造データを保存するかどうか聞いてきます。
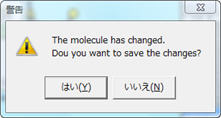
ここで「はい(Y)」を選ぶと、"temp.dat"という名前でMopac用の入力ファイルが保存されます。、
画面にDOS窓が現れて計算が始まります。(下の図は別な分子についての計算例です。)
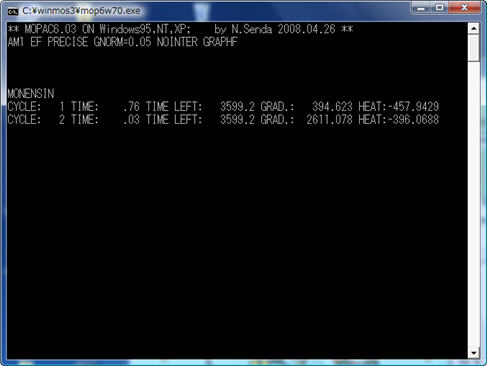
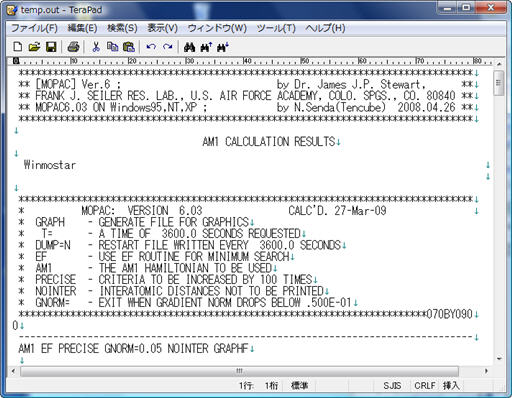
計算が終わると、計算結果が書かれたOutファイルが テキストエディタで開かれます。
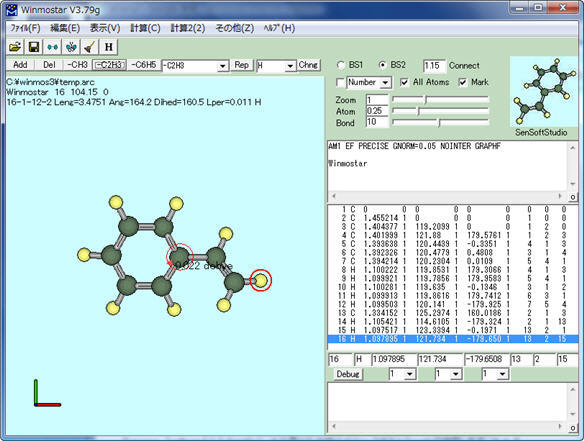
構造最適化計算が正常終了すると、最適化された構造が自動的に読み込まれ、分子表示ウィンドウに表示されます。
分子軌道の表示
今回の例のように、MOPACで計算を行う際にGRAPHキーワードが指定してある場合は、出力されたgptファイルまたはmgfファイルを読込むことで、Winmostarで分子軌道を表示することができます。
メニューから、[計算] - [分子軌道表示]を選び、gptファイルまたはmgfファイルを開きます。
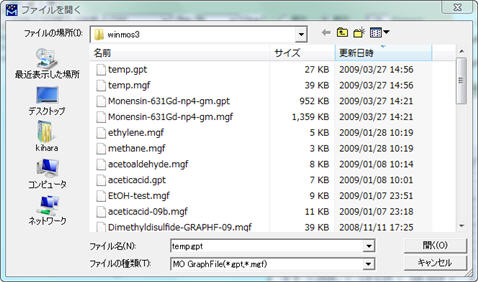
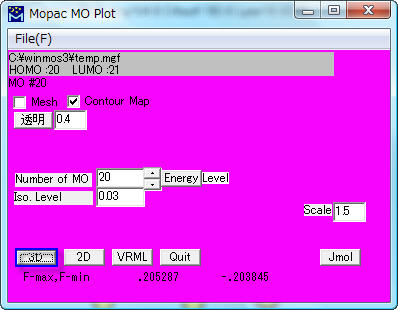
例えば、20番目の軌道であるHOMO(デフォールト)を指定して分子軌道を描きます。
分子軌道が描かれました。
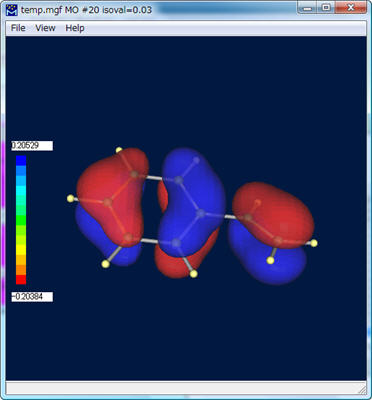
Winmostarでの分子軌道表示の表示については、[分子軌道表示]を参照してください。